
Myelofibrosis
Myelofibrosis (MF) is a chronic BCR::ABL1-negative myeloproliferative neoplasm (MPN) characterized by clonal myeloid proliferation, bone marrow fibrosis, extramedullary hematopoiesis, and progressive cytopenias.


Background
Myelofibrosis (MF) is a chronic BCR::ABL1-negative myeloproliferative neoplasm (MPN) characterized by clonal myeloid proliferation, bone marrow fibrosis, extramedullary hematopoiesis, and progressive cytopenias. It occurs either as primary MF (PMF) or secondary to antecedent PV or essential thrombocythemia (post-PV MF, post-ET MF). Median age at diagnosis is ~65 years. Hallmarks include constitutional symptoms, splenomegaly, progressive marrow failure, and leukemic transformation. Median survival is highly variable, ranging from >7 years in low-risk disease to <3 years in high-risk cases.
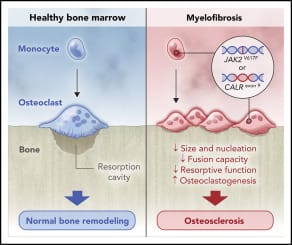
Pathophysiology
- Driver mutations:
- JAK2 V617F (~50%), CALR (~20–25%), MPL (~5–10%) activate JAK–STAT signaling
- ~5-10% are “triple-negative” (poorer prognosis)
- Extramedullary hematopoiesis arises as hematopoietic stem cells seed the spleen and liver
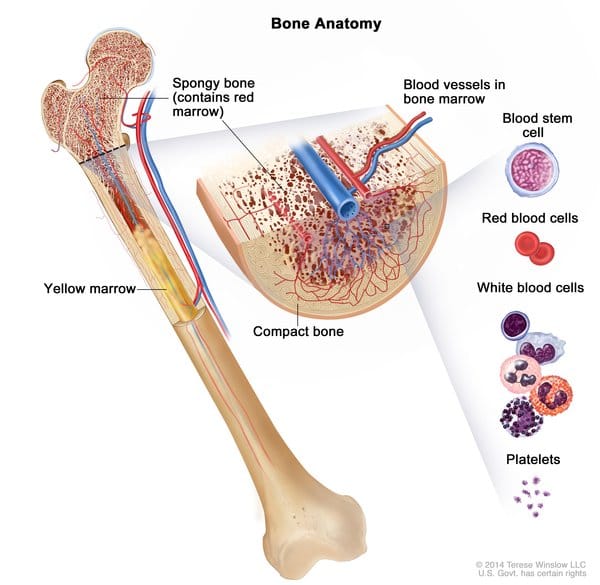
Risk Factors and Prognostic Scoring
Prognosis is heterogeneous; validated scoring systems include:
- IPSS/DIPSS/DIPSS-plus (age, symptoms, anemia, leukocytosis, blasts, transfusion dependence, platelets, karyotype)
- High-risk features: anemia (Hgb <10 g/dL), transfusion need, thrombocytopenia, circulating blasts ≥1%, constitutional symptoms, adverse mutations
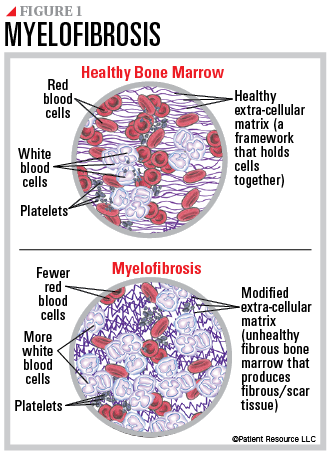
WHO Diagnostic Criteria
- Major (all 3 required)
- Bone marrow biopsy showing megakaryocytic proliferation/atypia with reticulin/collagen fibrosis (Grade ≥2)
- Exclusion of other myeloid neoplasms
- Presence of JAK2, CALR, or MPL mutation (or other clonal marker, absence of reactive fibrosis)
- Minor (≥1 required)
- Anemia not explained by comorbidity
- Leukocytosis ≥11×10^9/L
- Splenomegaly (clinical or imaging)
- Elevated LDH
- Leukoerythroblastosis
Work-up: H&P, symptoms, history of thrombosis or bleeding, CBC with smear, mutation testing (JAK2/CALR/MPL), bone marrow biopsy with reticulin/collagen grading, karyotype, imaging for splenomegaly.
Clinical Presentation
- Constitutional symptoms: fatigue, night sweats, weight loss, fever
- Splenomegaly: early satiety, abdominal discomfort, portal hypertension
- Cytopenias: anemia (most common), thrombocytopenia, leukopenia
- Other: bone pain, cachexia, extramedullary hematopoiesis (hepatomegaly, lymphadenopathy, pulmonary or CNS involvement)
- Complications: progression to acute myeloid leukemia
Treatment Goals
- Reduce symptom burden (constitutional and splenic)
- Improve quality of life and survival
- Manage cytopenias
- Prevent/slow progression to AML
Current Treatment Landscape
General/supportive care
- Red blood cell transfusions ± erythropoiesis-stimulating agents
- Iron chelation in transfusion-dependent younger patients
- Allopurinol for hyperuricemia; hydroxyurea for high WBC/platelet counts.
Allogeneic Hematopoietic Stem Cell Transplantation (all-HSCT)
- Considered for intermediate-2/high-risk disease in eligible patients
- Non-relapse mortality remains substantial; outcomes depend on age, comorbidities, donor type, and mutation profile
JAK inhibitors
- Ruxolitinib
- Fedratinib
- Pacritinib
- Momelotinib
Investigational Therapies
- Luspatercept
- Imetelstat
- Selinexor
- Bomedemstat
- Navtemadin
- Novel immunotherapies and fibrosis-targeting approaches are in development
Conclusion
Myelofibrosis is a complex MPN driven by JAK–STAT signaling and secondary clonal evolution, presenting with cytopenias, constitutional symptoms, and splenomegaly. Prognosis is heterogeneous and guided by integrated clinical, cytogenetic, and molecular scoring systems. JAK inhibitors—ruxolitinib, fedratinib, pacritinib, and momelotinib—form the backbone of symptomatic therapy.
The therapeutic landscape is rapidly expanding with agents targeting epigenetics, apoptosis, and fibrosis. The future of MF therapy will likely combine symptom control with true disease modification, aiming to extend survival and quality of life for patients across risk categories.
Key Points – Myelofibrosis
- Clonal MPN with marrow fibrosis, extramedullary hematopoiesis, cytopenias, splenomegaly, and constitutional symptoms
- Driver mutations: JAK2, CALR, MPL; triple-negative has worse outcomes
- Diagnosis (WHO 2022): requires BM fibrosis, clonal marker, and clinical/lab criteria
- Prognosis: scored with IPSS, DIPSS; highly heterogeneous survival
- Curative therapy: allo-HSCT in eligible, high-risk patients
- JAK inhibitors: Ruxolitinib, Fedratinib, Pacritinib, Momelotinib
- Emerging agents: luspatercept, imetelstat, selinexor, bomedemstat, navtemadin
- Future direction: move beyond symptom relief toward disease modification and survival improvement
References
- Tefferi A, Barbui T. Polycythemia vera and myelofibrosis: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023.
- Cervantes F, et al. DIPSS and prognostic scoring systems in MF. Blood. 2009–2018.
- Verstovsek S, et al. COMFORT-I/II: Ruxolitinib in MF. N Engl J Med. 2012.
- Harrison CN, et al. Fedratinib efficacy in MF. Lancet Haematol. 2017.
- Mascarenhas J, et al. Pacritinib in cytopenic MF. JAMA Oncol. 2022.
- Mesa RA, et al. Momelotinib in MF (MOMENTUM trial). Lancet. 2023.
- Kuykendall AT, et al. Emerging combination therapies (pelabresib, navitoclax). Clin Lymphoma Myeloma Leuk. 2024.
- Fenaux P, et al. Luspatercept in MPN-related anemia. Blood Adv. 2022.
- Tefferi A, et al. Imetelstat in refractory MF. J Clin Oncol. 2022.
*Information presented on RxTeach does not represent the opinion of any specific company, organization, or team other than the authors themselves. No patient-provider relationship is created.