
Polycythemia Vera
Polycythemia vera (PV) is a Philadelphia chromosome-negative (Ph-) myeloproliferative neoplasm (MPN) characterized by JAK-STAT pathway activation and trilineage myeloproliferation with a predominant increase in red blood cell mass.

Polycythemia vera (PV) is a Philadelphia chromosome-negative (Ph-) myeloproliferative neoplasm (MPN) characterized by JAK-STAT pathway activation and trilineage myeloproliferation with a predominant increase in red blood cell mass. Although rare (44-57 cases/100,000 prevalence), the disease poses great risk for patients if left untreated, including potential for thrombotic complications (arterial and venous, including splanchnic and cerebral venous sites). The median age at diagnosis is ~60 years, but late risks include potential evolution to post-PV myelofibrosis and acute myeloid leukemia.1
Pathophysiology
The disease is almost universally driven by somatic JAK2 mutations that confer cytokine-independent signaling:
- JAK2 V617F occurs in ~95% of PV, and JAK2 exon 12 mutations account for most of the remainder. These mutations activate JAK-STAT signaling downstream of the erythropoietin receptor, promoting erythroid proliferation and panmyelosis.
- Bone marrow shows hypercellularity with panmyelosis (erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes). Serum erythropoietin (EPO) is typically subnormal, reflecting negative feedback in primary erythrocytosis.
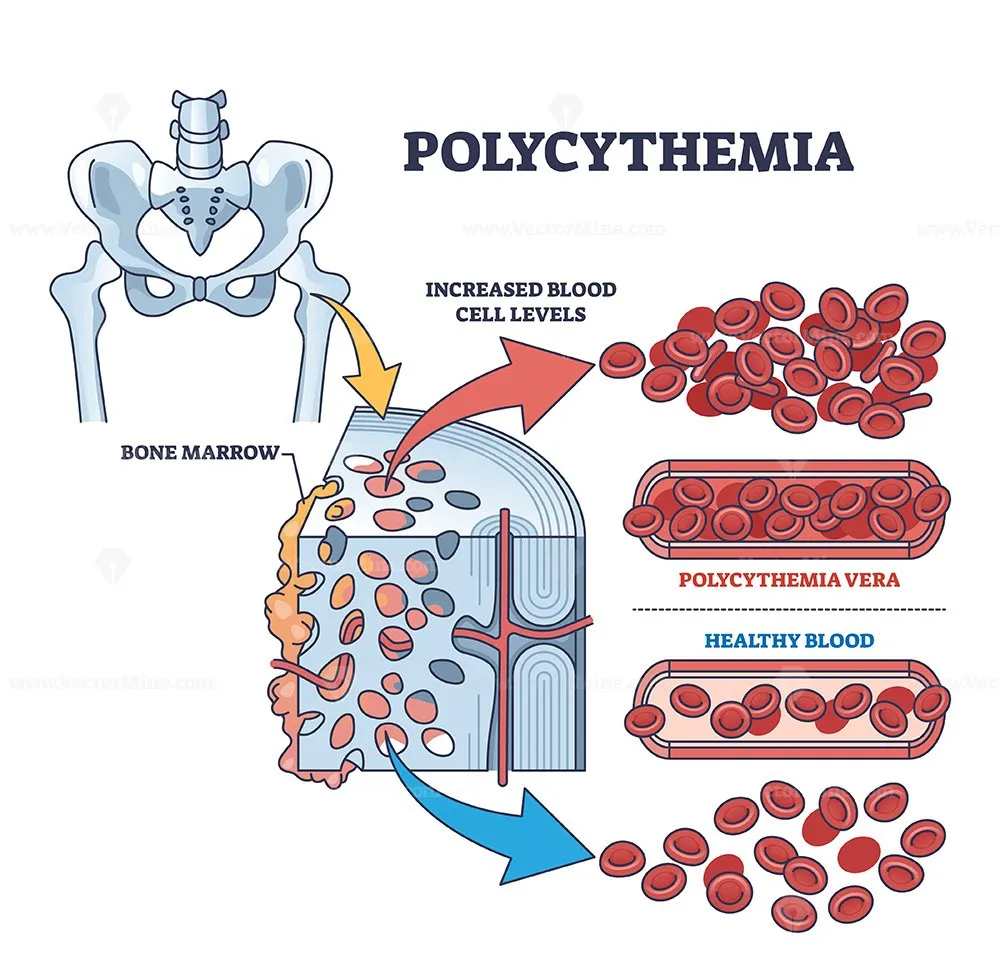
Risk Factors
Thrombosis risk is conventionally stratified as low risk (age <60 years and no prior thrombosis) versus high risk (age ≥60 and/or prior thrombosis). Additional modifiers (e.g., leukocytosis, cardiovascular risk factors) are often considered in practice. Two randomized trials anchor core prevention targets:
- ECLAP: low-dose aspirin reduced major vascular events versus placebo in PV without clear contraindications.2
- CYTO-PV: targeting hematocrit <45% significantly lowered cardiovascular death and major thrombosis compared with a 45–50% target. This <45% threshold underpins modern management.3
WHO Diagnostic Criteria
The WHO 5th edition (2022) framework (harmonized with 2016 thresholds) defines PV with three major and one minor criterion. Diagnosis requires all three major, or first two major + minor:1
Major
- Elevated hemoglobin/hematocrit or red cell mass:
- Hgb >16.5 g/dL (men) or >16.0 g/dL (women), or
- Hct >49% (men) or >48% (women), or
- Increased red cell mass
- Bone marrow biopsy:
- Hypercellular marrow with trilineage myeloproliferation and pleomorphic megakaryocytes
- Presence of a JAK2 mutation:
- V617F or exon 12
Minor
- Subnormal serum erythropoietin level
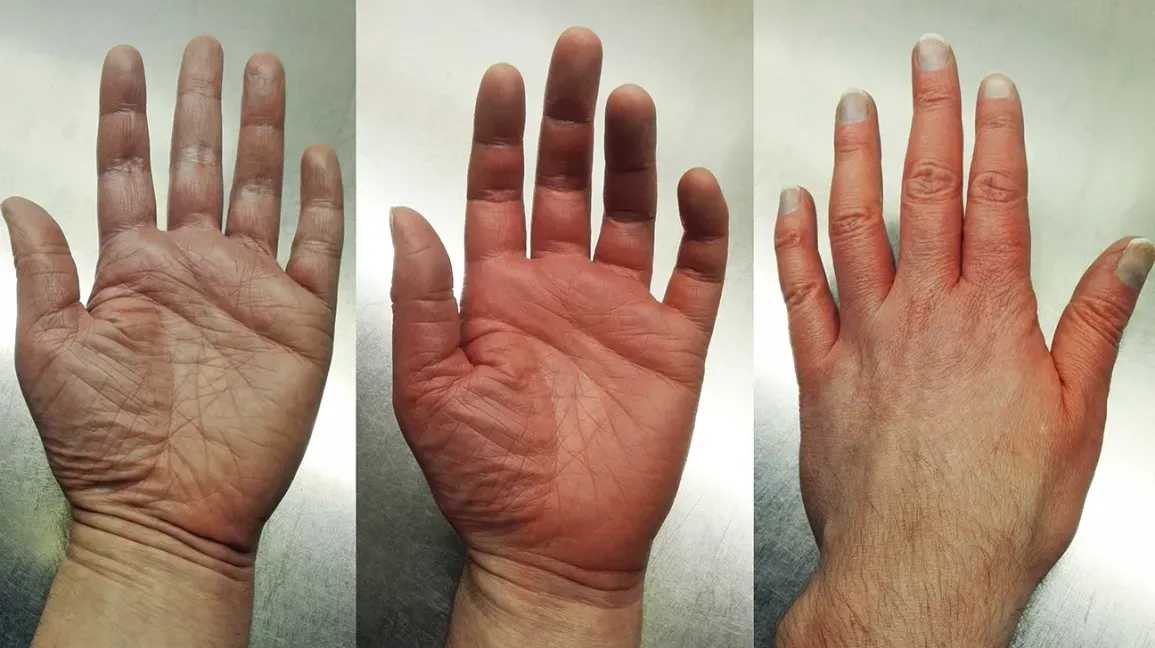
Clinical Presentation
Findings may include fatigue, pruritus, vasomotor disturbances, abdominal pain, early satiety, splenomegaly, and thrombosis. Iron deficiency from frequent phlebotomies may also occur. Over time, a subset progresses to post-PV myelofibrosis or AML, with increased risk for disease duration and certain therapy exposures.
Treatment Goals
- Prevent thrombosis/bleeding (aspirin, hematocrit control <45%, CV risk reduction)
- Control symptoms and splenomegaly
- Limit disease progression and minimize leukocytosis/thrombocytosis
Current Treatment Landscape
Foundational measures (all risk groups):
- Low-dose aspirin (typically 75–100 mg daily) if no contraindications
- Hold/avoid if extreme thrombocytosis with acquired von Willebrand syndrome2
- Phlebotomy to maintain Hct <45%3
- Optimize cardiovascular risk
- BP, lipids, smoking cessation, treat obstructive sleep apnea (OSA)1
Cytoreductive therapy
Indicated for high-risk patients and for selected low-risk patients with high symptom burden, progressive leukocytosis/thrombocytosis, frequent phlebotomy, or splenomegaly.
First-Line options
- Hydroxyurea (HU)
- Ropeginterferon alfa-2b (BESREMi)
- Binds to interferon alfa receptor, which modulates downstream signaling cascade through the activation of kinases (JAK, TYK, etc.) and activator of transcription (STAT) proteins. However, the actions involved in the therapeutic effects of interferon alfa in polycythemia vera are not fully elucidated.4
Second-Line/HU-Resistant or Intolerant
- Ruxolitinib (JAK1/2 inhibitor)
- Inhibits JAK1 and JAK2, which mediate the signaling of a number of cytokines and growth factors that are important for hematopoiesis and immune function.5
Agent selection often hinges on patient age, symptom profile, comorbidities, pregnancy considerations, and preferences regarding goals (disease modification, symptom/spleen control, route of administration, etc.). Shared decision-making and guideline-concordant monitoring is crucial.
Investigational/Emerging Therapies
- Rusfertide (PTG-300) is a hepcidin mimetic that restricts iron availability and erythropoiesis, and it has shown marked reductions in phlebotomy needs and hematocrit control in clinical trials.7
- Givinostat is an HDAC inhibitor that leads to the acetylation of histones and potential downregulation of JAK2.
Key Points
- Rare Ph- MPN with trilineage myeloproliferation & increased Hct/red cell mass
- Driven by JAK2 mutations (V617F ~95%) resulting in JAK-STAT signaling/panmyelosis
- Symptoms include fatigue, pruritus, splenomegaly, with a high concern for thrombosis
- Universal treatment includes low-dose aspirin (if safe) and Hct <45% through phlebotomy ± cytoreduction
- Cytoreduction for high-risk (>60 years old and/or thrombosis): HU or ropeginterferon alfa-2b
- If HU fails (resistance/intolerance), ruxolitinib is evidence-based for hematocrit, spleen, and symptom control and can reduce thrombotic events
- Monitor erythrocytes, hemoglobin, hematocrit, leukocytosis, and platelets; control CV risks; reassess symptom burden; be vigilant for myelofibrotic transformation
Conclusion
Polycythemia vera is a chronic, JAK2-driven MPN that carries substantial risks of thrombosis, disease-related symptoms, and eventual potential progression to myelofibrosis or acute leukemia. Advances in understanding its molecular pathophysiology have shaped modern diagnostic frameworks, anchored by JAK2 mutation testing, bone marrow morphology, and WHO-defined thresholds for erythrocytosis. Evidence from pivotal trials has firmly established hematocrit control (<45%) and low-dose aspirin as the foundation of care, while risk-adapted cytoreductive therapy with hydroxyurea or peginterferon alfa-2b provides a potential for further protection against thrombosis and disease progression. Ruxolitinib offers an effective option in patients resistant or intolerant to hydroxyurea, with potential benefits in hematocrit control, splenomegaly, and symptom burden.
The therapeutic landscape is also evolving, with agents such as rusfertide and givinostat showing unique mechanisms of action. Optimal care therefore requires vigilant monitoring, thrombotic risk reduction, and individualized therapies that meet both patient and provider goals of care.
References
- Tefferi A. Am J Hematol. 2023—2024 updates on diagnosis, risk stratification, and treatment.
- Landolfi R, et al. ECLAP randomized trial. N Engl J Med. 2004.
- Marchioli R, et al. CYTO-PV randomized trial. N Engl J Med. 2013.
- FDA BESREMi label (ropeginterferon alfa-2b-njft); PROUD-PV/CONTINUATION-PV long-term results (Leukemia 2023).
- Vannucchi AM, et al. RESPONSE. N Engl J Med. 2015; Verstovsek S, et al. RESPONSE-2/Haematologica; MAJIC-PV.
- Protagonist Therapeutics (rusfertide VERIFY topline, 2025).
- Chifotides HT, et al. Givinostat: an emerging treatment for polycythemia vera. Expert Opin Investig Drugs. 2021.
*Information presented on RxTeach does not represent the opinion of any specific company, organization, or team other than the authors themselves. No patient-provider relationship is created.